Fibrolamellar carcinoma (FLC), a rare liver cancer in young people, is caused by a gene fusion due to a chromosome 19 deletion, discovered by Sanford M. Simon’s lab in 2014. Recent research from the same lab revealed that the cancer is driven by the overproduction of the PKA protein, not by structural changes in the kinase.
Recent research has redefined the cause of fibrolamellar carcinoma, a rare liver cancer, showing that it stems from the overproduction of the PKA protein rather than a gene fusion, opening up potential new treatment avenues.
It’s been enormously difficult to pinpoint the cause of many cancers—and many seem to have more than one origin. Fibrolamellar carcinoma (FLC), however, is one that scientists thought they had nailed down.
A rare and currently incurable disease that attacks the liver of children, adolescents, and young adults, FLC is caused when a small deletion in chromosome 19 causes the fusion of two genes, a discovery made in 2014 in the lab of Rockefeller’s Sanford M. Simon, whose own then-teenage daughter, Elana, had not only been diagnosed with liver disease a few years before, but was a lead on the team that found the fusion.
One gene is DNAJB1, which produces heat-shock proteins that encourage cell homeostasis, and the other is PRKACA, the generator of the catalytic subunit of protein kinase A (PKA), which is key to cellular metabolic function. Alterations in kinases, which modify many other molecules, have been implicated in driving many cancers.
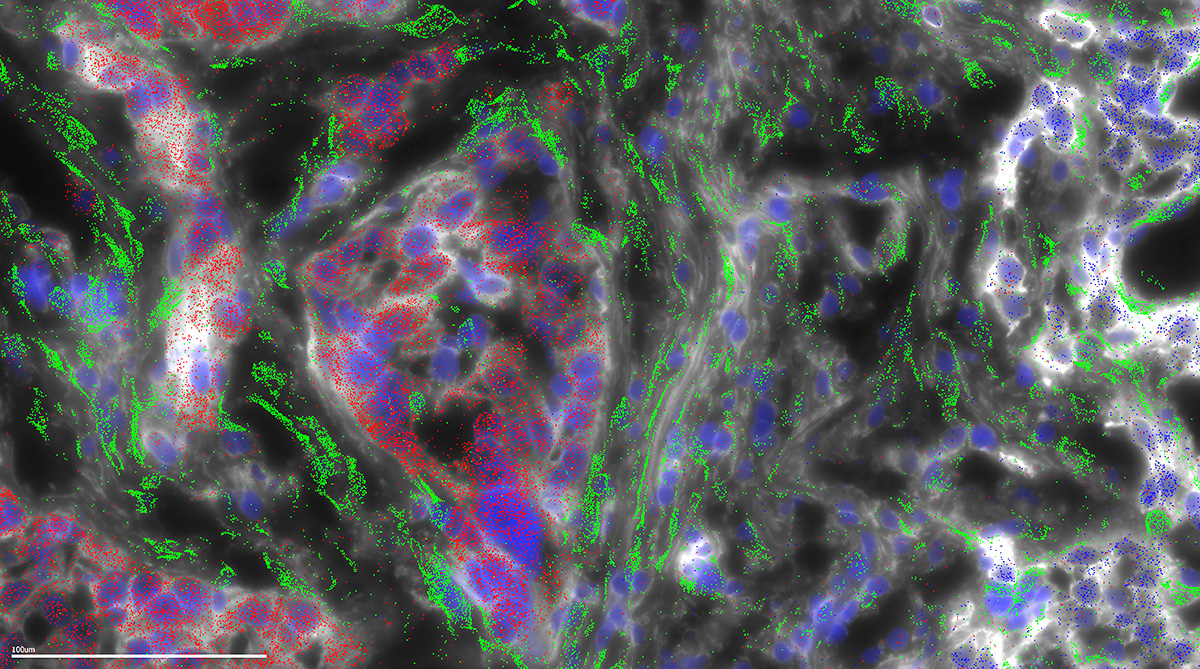
Fibrolamellar carcinoma (FLC) gets its name from the fibrous bands of collagen that run through the tumor. In the current study, microscopy was used to map where the different genes are expressed in a FLC tumor. This microscopy shows that the collagen is being generated not by the FLC tumor cells (red), but instead in the wispy host cells (green) called stellate cells, in response to the FLC cells. Credit: Laboratory of Cellular Biophysics at The Rockefeller University
For the past decade, it was thought that this fusion created a Frankenstein-type change in PKA so that it wreaked havoc in the cell. Now researchers in Simon’s lab have made a surprising discovery: the fusion protein behaves just like a normal kinase. But cells containing an addition to their catalytic subunit generate the kinase in excessive amounts—which is the real culprit.
“It’s actually the overexpression of a protein called PKA that causes the cancer,” says first author Mahsa Shirani, a postdoctoral associate in the Laboratory of Cellular Biophysics, headed by Simon. “These findings have the potential to both reveal the pathways of a broad range of cancers and offer new treatment possibilities.”
The researchers published their results in Cancer Research.
A lack of inhibition
Shirani’s research has been aimed at a deeper understanding of the fused gene’s mechanics since her time as a Ph.D. student and teaching assistant in the lab of New Mexico State University biochemist Barbara Lyons, whose research into FLC was driven by her own son’s diagnosis with the disease. Like Elana Simon and numerous other patients, her son, Jackson Clark, put his life on hold to research the disease in the Simon lab. His first article from the lab was published last year. Clark passed away from FLC.
For the current study, Shirani analyzed tumor tissue samples from FLC patients using mass spectrometry, biochemistry, and immunofluorescence to quantify the level of the protein in patients’ tumor tissue. She also compared them to normal liver tissue.
Digging deeper, she found that the tumor cells have a molecular imbalance: an increased amount of catalytic proteins exceeds the number of inhibitory ones that normally tamp down and localize the former. This excess has two profound effects on the cell. One is that PKA activity amps up, unchecked. The other is that PKA is now free to move around the cell, wreaking havoc in spots that it usually cannot access, including the nucleus.
Shirani’s results indicate that the fact that the active catalytic subunit overrules its inhibitory components is what’s important, not a structural change in the kinase itself.
In testing this theory, the researchers found that they could recreate the disease in human liver cells just by increasing the amount of the normal kinase. They also found that some patients had a completely different gene fused to the front end of the same kinase, PRKACA, indicating that the actual cause of disease couldn’t be attributed to the extra piece added to the kinase.
“We showed that it doesn’t matter what you have fused to the PRKACA gene. It could be DNAJB1 or ATP1B1, or it could be nothing at all—just PRKACA that has a high protein expression,” she says. “Each situation leads to the same phenotype of cancer.”
The researchers corroborated their findings using a unique tool at their disposal. For the past decade, the Simon Lab has operated the Fibrolamellar Tissue Repository. When the researchers went back through their samples, they came across four patients who had what looked like fibrolamellar but did not have a fusion to PRKACA. Instead, the only alteration they found was a loss of the inhibitory protein, providing more evidence that the amount of catalytic subunit relative to its regulatory components was a key factor in disease formation.
Treatment horizon
The findings could potentially lead to the first therapeutic treatments for FLC beyond the surgical removal of tumors, says Shirani. (Treatments available to people with common liver cancer are useless for FLC, which has a completely different molecular profile.)
One idea is to locate binding sites on the DNAJB1 protein to which a drug inhibitor could bind. Another is tamping down the expression of PKA. The lab is currently investigating both possibilities.
The latter approach could have potential beyond FLC, Shirani says, because PKA dysregulation is connected to many other diseases. For example, the adrenal tumor that causes Cushing Syndrome is the result of a mutation in the very same catalytic subunit, PRKACA. (That discovery was made by Rockefeller President Richard P. Lifton in 2014.)
As with a potential FLC treatment, the key would be to interfere with signaling processes downstream of PKA’s production, before the geyser of protein production disrupts the cell.
Shirani also suggests that measuring protein levels produced by mutated genes may be a first step to better understanding any number of cancers: “Perhaps the increased level, or location, of proteins is itself the cause.”
The findings may also illuminate pathogenesis of disease in general—one of the many important reasons for investigating rare diseases, which are often viewed as insignificant since they affect so few people, says Simon.
“There are so many good reasons to study them,” he says. “Many rare diseases are very precisely characterized, making it possible to make rapid progress, and those results can often be generalized to common diseases. For example, we learned about the concept of ‘tumor suppressor’ from studying the rare childhood cancer retinoblastoma.”
“I also think that as we more precisely define diseases, we find that many that were thought to be single diseases are actually collections of different rare diseases that share some common characteristic or mechanism,” he adds.
Reference: “Increased Protein Kinase A Activity Induces Fibrolamellar Hepatocellular Carcinoma Features Independent of DNAJB1” by Mahsa Shirani, Solomon Levin, Bassem Shebl, David Requena, Tova M. Finkelstein, Daniel S. Johnson, Denise Ng, Gadi Lalazar, Søren Heissel, Peter Hojrup, Henrik Molina, Ype P. de Jong, Charles M. Rice, Aatur D. Singhi, Michael S. Torbenson, Philip Coffino, Barbara Lyons and Sanford M. Simon, 18 June 2024, Cancer Research.
DOI: 10.1158/0008-5472.CAN-23-4110